import {Plot} from "@mkfreeman/plot-tooltip"
import {map} from "@martien/ramda"
papers_r = FileAttachment("../data/all_papers.json").json()
n_columns = Object.entries(papers_r["doi"]).length
index_array = Array.from({length: n_columns}, (_, i) => i)
papers = map(i => map(x => x[i], papers_r), index_array)
function displayTable(papers_) {
return Inputs.table(papers_, {
columns: [
"doi",
"title",
"journal.name",
"journal.volume",
"year",
],
header: {
"journal.name": "journal",
"journal.volume": "volume",
},
format: {
doi: doi => htl.html`<a href=https://doi.org/${doi} target=_blank>${doi}</a>`,
title: title => htl.html`${title}`,
"journal.volume": volume => htl.html`<b>${volume}</b>`,
year: year => htl.html`${year}`,
},
width: {
"title": 10
},
layout: "auto",
})
}Research topics
Overview
Our research focuses on the development and application of multiscale molecular simulations methods for soft-condensed-matter materials. We are particularly invested in using multiscale modeling to explore chemical compound space. The group develops methodologies to accelerate compound-space exploration by means of high-throughput molecular dynamics simulations. Transferable coarse-grained models have the capability to reduce the size of chemical space—a property we leverage to more easily navigate the thermodynamic properties of a large subset of chemical compounds. Coarse-graining also eases the identification of structure–property relationships and design rules for molecular discovery. Other activities include method development of coarse-grained models; machine learning for soft matter; non-equilibrium dynamical reweighting; force-field development; polymer, protein, and phospholipid membrane simulations.
Coarse-graining for molecular discovery
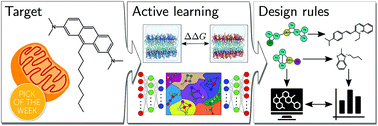
Coarse-grained (CG) molecular simulations are used for molecular discovery of small molecule probes that specifically bind to cardiolipin. We combine rigorous free-energy calculations, selectivity prediction inside a low-dimensional chemical-space embedding, and Bayesian optimization to identify physicochemical design rules. Said design rules are used to further filter a chemical vendor database. Out of 20 compounds tested, we identify three compounds selective in vitro and even one in vivo.
Machine learning for soft-matter materials
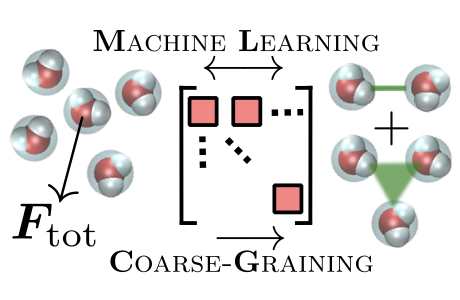
We explore the links between machine learning (ML) and multiscale molecular modeling. Emphasis is placed on tailoring ML models to problem at hand, may that be in the representation of the architecture itself. Kernel-ridge regression, used for small datasets, eases the (sometimes analytical) tailoring of physical properties. Deep neural networks offer more flexibility, expressivity.
Non-equilibrium dynamical reweighting
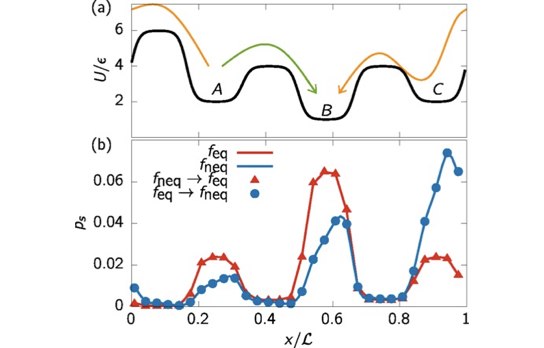
We extend the concept of statistical reweighting to non-equilibrium steady states. We use stochastic thermodynamics to compute the entropy production along microtrajectories and reweigh them using a Maximum Caliber approach. Extension to collective variables is presented.
Deep backmapping
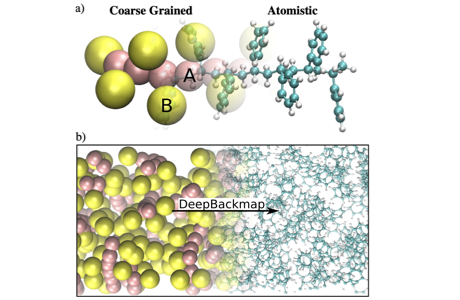
We generate condensed molecular structures as a refinement—backmapping—of a coarse-grained structure. We apply generative adversarial networks (GAN) conditional on the coarse-grained configuration to upscale to atomistic structure. Sampled configurations accurately target the atomistic Boltzmann distribution, and do not require pre-equilibration steps. Remarkable transferability is found across temperature for a polymer melt. Applications to chemical transferability are also investigated.
Kinetic properties of coarse-grained models
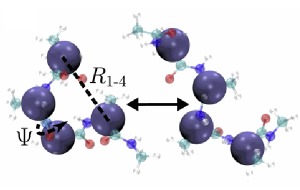
We analyze and improve the kinetic properties of coarse-grained simulations. Methodologies include Markov state models biased with external information; Conformational surface hopping for improved reconstruction of the potential of mean force.
High-throughput screening of thermodynamic properties
We use coarse-graining (CG) to emulate a high-throughput screening experiment for free-energy calculations. We start from the CG Martini model, and tailor the bead parametrization to explore the chemical space of small molecules. Applications tailored to phopholipid membranes.
Machine learning of non-covalent interactions

We combine physics-based potentials and machine learning for accurate and transferable non-covalent interactions. Physics-based models include atomic multipoles and many-body dispersion. The coefficients are learned by kernel-ridge regression across conformations and composition of small organic molecules.
Static atomic multipole electrostatics in condensed-phase simulations
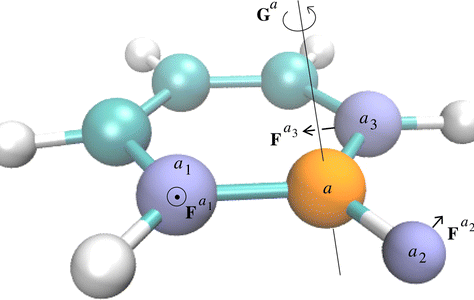
Static atomic multipoles provide an improved description of the electrostatic potential of the system. Molecular dynamics simulations of condensed-phase systems demonstrate enhanced equilibrium and dynamical quantities: e.g., free energy of hydration and 2D infra-red spectroscopy.
Structure formation in peptide coarse-graining
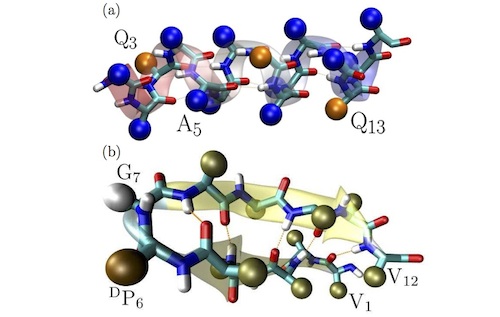
Using a top-down coarse-grained peptide model, we study secondary and tertiary structure formation in different environments and scenarios: alpha-helix vs. beta-sheet folding, microcanonical analysis of folding, structural alignment of capsid interfaces, and peptide-membrane interactions.